Le encefalopatie spongiformi (malattie da prioni) sono quelle malattie in cui sono coinvolte nello sviluppo forme patologiche di proteine prioniche. Conosciamo sempre di più le malattie da prioni, ma gli aspetti chiave sono ancora sconosciuti: attualmente la medicina non ha i mezzi per curare i pazienti da queste malattie.
Le encefalopatie spongiformi, o malattie da prioni, possono svilupparsi durante la vita, mentre altre derivano da mutazioni genetiche ereditarie presenti dalla nascita. All'interno di questo gruppo, ci sono diverse entità che si verificano negli esseri umani, esempi sono la malattia di Creutzfeldt-Jakob o l'insonnia familiare fatale.
Le malattie da prioni sono state molto misteriose per molto tempo. A differenza di altri patogeni, come batteri, virus o funghi, non contengono acido nucleico: i prioni sono fatti solo di proteine. La teoria delle malattie da prioni è stata scoperta da S. Prusiner, questa scoperta è stata molto apprezzata dalla comunità scientifica - nel 1997 il ricercatore ha ricevuto il Premio Nobel per la medicina. Sebbene siano passati relativamente molti anni da quando è nato il concetto di prione, alcuni scienziati ritengono ancora che sia incompleto e stanno studiando ulteriormente la natura di queste condizioni: alcuni dei fattori responsabili delle encefalopatie spongiformi sono stati ora confermati.
Malattie da prioni: cause
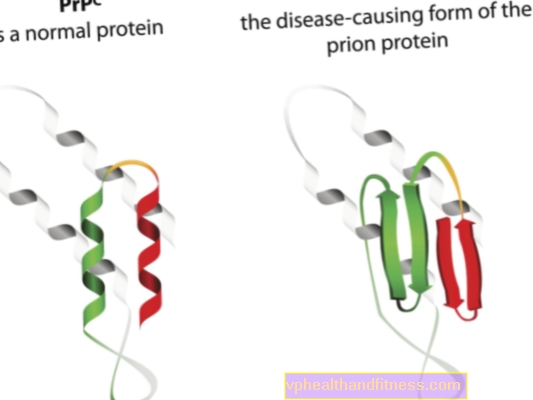
L'eziologia delle malattie da prioni è correlata alla trasformazione delle normali proteine prioniche in forme patogene e patogene. I prioni sono molecole proteiche che si trovano nel corpo di ogni essere umano. La loro funzione non è ancora del tutto chiara, ma è noto che in condizioni normali, le proteine prioniche non danneggiano il corpo. Tuttavia, quando i prioni cambiano la loro struttura e diventano particelle patogene, si sviluppa una delle numerose encefalopatie spongiformi. I prioni presenti naturalmente nel corpo sono indicati come PRPC, mentre le forme anormali sono indicate come PRPSC. Questi ultimi sono un problema serio non solo perché possono accumularsi nel tessuto nervoso sotto forma di depositi e generarne il danno, ma anche perché hanno la capacità di trasformare i normali prioni in una forma malformata (in poche parole, PRPSC può "infettare" proteine normali con il suo potenziale patogeno).
Leggi anche: Malattia di Huntington (corea di Huntington): cause, sintomi, trattamento Tremori muscolari - cause. Cosa significa tremore muscolare? Malattie che uccidono più velocemente: SHOCK, EBOLA, DAMN, ATTACK, EMERGENCY [GALE ...Fondamentalmente, ci sono 3 cause di encefalopatie spongiformi:
- sporadico (la mutazione patogena si verifica nelle cellule somatiche, si verifica durante la vita del paziente),
- famiglia (derivante dal peso delle mutazioni ereditate dai genitori),
- Passaggio (correlato all'introduzione di prioni patogeni nel corpo umano, ad esempio attraverso preparati di ormone della crescita contaminati da queste particelle o trapianto di cornea da una persona affetta da qualche encefalopatia spongiforme).
Encefalopatie spongiformi: malattia di Creutzfeldt-Jakob
La malattia di Creutzfeldt-Jakob (CJD) è stata descritta per la prima volta all'inizio degli anni '20. Esistono 4 tipi di malattia:
- MCJ sporadica (la più comune, che rappresenta fino al 9/10 di tutti i casi di MCJ)
- città natale di CJD
- sopraffatto da CJD
- variante di CJD
Il quadro clinico nel corso di varie varianti della malattia di Creutzfeldt-Jakob può essere variabile. I disturbi più comuni nel corso di questo gruppo di encefalopatie spongiformi sono:
- disturbi della demenza (compreso il progressivo deterioramento della memoria, dell'attenzione e della concentrazione)
- mioclono (movimenti involontari come spasmi muscolari improvvisi)
- disfunzione cerebellare (manifestata, ad esempio, da disturbi dell'equilibrio)
- visione offuscata
- sintomi piramidali ed extrapiramidali
Nel corso delle varianti CJD, possono comparire anche disturbi mentali (ad esempio ansia, umore depresso), dolore e altri movimenti involontari diversi da quelli sopra menzionati.
La prognosi per la malattia di Creutzfeldt-Jakob è infausta - ad esempio, nei pazienti con MCJ sporadica, ci vogliono in media dai quattro ai cinque mesi dall'inizio dei sintomi della malattia alla morte.
Encefalopatie spongiformi: sindrome di Gerstmann-Straussler-Scheinker
La sindrome di Gerstmann-Straussler-Scheinker (GSS) di solito è familiare ed è causata da una mutazione ereditaria nel gene PRNP. È considerata l'encefalopatia spongiforme a più lenta progressione. Il team GSS comprende:
- atassia spinocerebellare
- disartria
- disturbi della demenza
- disturbi della deglutizione
- nistagmo
- aumento della tensione muscolare
I pazienti con diagnosi di GSS hanno un periodo di tempo variabile e in alcuni pazienti la morte si verifica più di 10 anni dopo l'esordio.
Encefalopatie spongiformi: insonnia familiare fatale
L'insonnia familiare letale è una malattia da prioni causata da mutazioni nel gene PRNP. La malattia è estremamente rara ed è stata finora diagnosticata in 28 famiglie in tutto il mondo. Nel corso dell'insonnia familiare fatale, il primo sintomo è l'incapacità di dormire. Questo problema provoca disturbi d'ansia e il paziente avverte allucinazioni. L'effetto della costante mancanza di riposo notturno è la disfunzione del sistema autonomo (compresi i cambiamenti nella funzione cardiaca, sudorazione e disturbi del sistema digestivo), c'è anche una progressiva diminuzione del peso corporeo. Negli stadi più avanzati dell'insonnia familiare fatale, compaiono disturbi ormonali e nel corso della malattia si verificano sintomi di demenza.
La prognosi per l'insonnia familiare fatale, come per altre encefalopatie spongiformi, è infausta: i pazienti di solito muoiono entro tre anni dall'esordio.
Encefalopatie spongiformi: prionopatia con suscettibilità variabile alla proteasi
Il verificarsi delle encefalopatie spongiformi discusse è principalmente correlato alle mutazioni nel gene PRNP. Tuttavia, queste mutazioni riguardano diversi codoni di questo gene e pertanto si distinguono diverse malattie da prioni differenti. Un'unità descritta relativamente di recente (nel 2008) è la prionopatia con suscettibilità variabile alla proteasi. Le persone che soffrono di questa malattia portano mutazioni in ben tre codoni del gene PRNP.
Nella prionopatia con sensibilità alla proteasi variabile, i pazienti sperimentano:
- decadimento cognitivo
- estrema gravità dei disturbi psichiatrici: possono essere euforia e agitazione, ma anche significativa apatia
- disartria
- afasia (disturbi delle funzioni linguistiche)
La durata media della malattia in questa prionopatia è inferiore a 4 anni.
Encefalopatie spongiformi: kuru
Il kuru è ora considerato una malattia che praticamente non esiste più: è stato trovato in rappresentanti di tribù della Papua Nuova Guinea, che praticavano comportamenti cannibali. Il sintomo dominante di questa encefalopatia spongiforme è l'atassia cerebellare progressiva. Può essere accompagnato da movimenti involontari (principalmente sotto forma di corea, tremori e atetosi), nonché incontinenza urinaria e fecale. I pazienti in kuru sperimentano anche significativi sbalzi d'umore, sviluppano riflessi primitivi (ad esempio succhiare). Un problema piuttosto caratteristico nel caso di questa malattia da prioni sono gli attacchi forzati di pianto o risate - a causa di quest'ultimo fenomeno, il kuru è a volte indicato come "morte per risata".
Encefalopatie spongiformi: diagnosi
Le malattie da prioni possono essere sospettate sulla base dei sintomi del paziente. Tuttavia, sono piuttosto aspecifici, poiché possono anche manifestarsi nel corso di una serie di altre malattie non correlate ai prioni. Per questo motivo, nella diagnosi delle encefalopatie spongiformi vengono utilizzati anche i seguenti:
- test di imaging (ad esempio la risonanza magnetica, che consente di rilevare i cambiamenti legati alla degenerazione del cervello da parte delle proteine prioniche),
- test di laboratorio (come la valutazione delle concentrazioni di proteine nel liquido cerebrospinale, ad esempio proteine MAP-tau, S-100 o 14-3-3),
- test genetici (per rilevare la presenza di mutazioni nel paziente),
- test immunoistochimici (utilizzando anticorpi contro le proteine prioniche).
La diagnosi può essere confermata anche dall'autopsia cerebrale, in cui è possibile riscontrare alterazioni caratteristiche delle encefalopatie spongiformi. Queste possono essere lesioni spugnose, variamente distribuite e con una struttura diversa (a seconda dell'entità specifica della malattia), placche amiloidi e difetti neuronali.
Encefalopatie spongiformi: trattamento
Le malattie da prioni sono attualmente incurabili: nonostante i numerosi studi in corso da molti anni, la medicina non ha ancora farmaci che potrebbero rallentare o inibire completamente il loro progresso. Il trattamento sintomatico è utilizzato nei pazienti con encefalopatie spongiformi, il cui scopo è alleviare l'intensità dei sintomi e migliorare il più possibile la loro qualità di vita.
Tuttavia, il lavoro sul trattamento delle encefalopatie spongiformi è ancora in corso. Gli scienziati stanno cercando di utilizzare vari metodi: il primo esempio è la terapia genica. Influirebbero sugli acidi nucleici e sulle mutazioni presenti nella loro struttura: lo scopo dell'applicazione della terapia genica sarebbe neutralizzare gli errori nel codice genetico. Un approccio diverso è alla base della terapia immunitaria: sono in corso lavori per creare anticorpi il cui ruolo sarebbe quello di eliminare i prioni patogeni. Un altro metodo che vede il potenziale per combattere le encefalopatie spongiformi è il trattamento con l'utilizzo di molecole proteiche sintetizzate, che, una volta introdotte nel corpo del paziente, neutralizzerebbero le proteine patologiche.
Articolo consigliato:
Encefalopatie: cause, tipi e sintomi