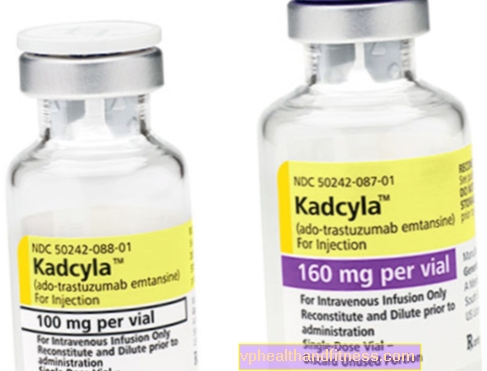
1 flaconcino contiene 100 mg (160 mg) di polvere per la ricostituzione 5 ml (8 ml) di trastuzumab emtansine concentrato a una concentrazione di 20 mg / ml per infusione dopo la ricostituzione.
| Nome | Contenuto della confezione | Il principio attivo | Prezzo 100% | Ultima modifica |
| Kadcyla | 1 flaconcino, polvere per la preparazione soluzione finale a inf. | Trastuzumab emtansine | 2019-04-05 |
Azione
Farmaco antitumorale. Trastuzumab emtansine è un coniugato farmaco-anticorpo che contiene trastuzumab, un anticorpo monoclonale IgG1 anti-HER2 umanizzato, legato in modo covalente a DM1 (derivato della maytansina, inibitore dei microtubuli) tramite un collegamento tioetere MCC stabile (4- cicloesano-1-carbossilato) . Emtansine è il complesso MCC-DM1. La coniugazione di DM1 con trastuzumab determina l'azione selettiva di farmaci citotossici contro le cellule tumorali sovraesprimenti HER2, aumentando così la concentrazione intracellulare di DM1 direttamente nelle cellule tumorali. Dopo il legame con HER2, trastuzumab emtansine è internalizzazione mediata dal recettore seguita da degradazione lisosomiale, rilasciando cataboliti contenenti DM1 (principalmente lisina-MCC-DM1). Il meccanismo d'azione di trastuzumab emtansine è mediato dall'attività di trastuzumab e DM1. Il trastuzumab emtansine, come il trastuzumab, si lega al dominio IV del dominio extracellulare (ECD) del recettore, nonché ai recettori Fcγ e al complemento C1q. Inoltre, inibisce l'attività del dominio ECD del recettore HER2, inibisce la segnalazione della via della fosfatidilinositolo 3-chinasi (PI3-K) e media la citotossicità cellulare dipendente da anticorpi (ADCC) nelle cellule di cancro al seno umano che sovraesprimono HER2. DM1 si lega alla tubulina. Inibendo la polimerizzazione della tubulina, sia DM1 che trastuzumab emtansine provocano l'arresto delle cellule nella fase G2 / M del ciclo cellulare, portando infine alla morte cellulare per apoptosi. Il linker MCC riduce il rilascio sistemico di DM1 e aumenta la sua concentrazione nel sito target. Trastuzumab emtansine viene deconiugato e quindi catabolizzato mediante proteolisi nei lisosomi cellulari. DM1 è metabolizzato principalmente dal CYP3A4 e in misura minore dal CYP3A5. Il T0,5 di trastuzumab è di circa 4 giorni. Non si è verificato alcun accumulo di trastuzumab emtansine a seguito di più infusioni endovenose somministrate a un intervallo di 3 settimane. L'età non ha avuto effetti sulla farmacocinetica di trastuzumab emtansine.
Dosaggio
Per via endovenosa. Il preparato deve essere prescritto da un medico e somministrato sotto la supervisione di operatori sanitari esperti nel trattamento di pazienti oncologici. I pazienti che ricevono trastuzumab emtansine dovrebbero avere un cancro HER2 positivo - punteggio immunoistochimico (IHC) 3+ o rapporto ≥2 all'ibridazione in situ (ISH). I test devono essere eseguiti utilizzando test diagnostici in vitro (IVD) con marchio CE. Se il test CE IVD non è disponibile, il test deve essere eseguito con un altro test convalidato. Per evitare errori medici, è importante controllare l'etichetta del flaconcino per assicurarsi che il medicinale che viene preparato e somministrato sia Kadcyla (trastuzumab emtansine) e non Herceptin (trastuzumab). La dose raccomandata è di 3,6 mg / kg di peso corporeo. somministrato come infusione endovenosa ogni 3 settimane (ciclo di 21 giorni). I pazienti devono essere trattati fino alla progressione del tumore o al raggiungimento di una tossicità inaccettabile. La dose iniziale deve essere somministrata come infusione endovenosa di 90 minuti. Il sito di iniezione deve essere attentamente monitorato poiché il farmaco può penetrare per via sottocutanea durante la somministrazione. Se la precedente infusione è stata ben tollerata, le dosi successive possono essere somministrate come infusione di 30 minuti. I pazienti devono essere osservati durante l'infusione e per almeno 30 minuti successivamente. La velocità di infusione deve essere ridotta o interrotta se il paziente manifesta sintomi correlati all'infusione. Trastuzumab emtansine deve essere interrotto in caso di reazioni all'infusione pericolose per la vita. Devono essere disponibili per l'uso immediato farmaci per reazioni allergiche / anafilattiche all'infusione, nonché attrezzature di emergenza. Se una dose pianificata viene dimenticata, deve essere somministrata il prima possibile; non aspettare fino al ciclo successivo. Il programma di dosaggio deve essere regolato per mantenere l'intervallo di somministrazione di 3 settimane. La dose successiva deve essere somministrata secondo le raccomandazioni sul dosaggio. La gestione delle reazioni avverse sintomatiche può includere l'interruzione periodica, la riduzione della dose o l'interruzione del trattamento. Schema di riduzione della dose (dose iniziale 3,6 mg / kg): prima riduzione della dose 3 mg / kg; seconda riduzione della dose 2,4 mg / kg di peso corporeo; se è necessaria un'ulteriore riduzione della dose, il trattamento deve essere interrotto. Linee guida per la modifica della dose per aumenti di AST e ALT: Grado 2 (da> 2,5 a ≤5 x ULN) - non è necessario alcun aggiustamento della dose; grado 3 (da> 5 a ≤20 x ULN) - utilizzare il preparato se AST / ALT diminuisce al grado ≤ 2 (da> 2,5 a 20 x ULN) - interrompere il trattamento. Regole di modifica della dose per l'iperbilirubinemia: grado 2 (da> 1,5 a ≤3 x ULN) - utilizzare la preparazione quando il livello di bilirubina è sceso al grado ≤1. (> ULN a 1,5 x ULN), non è necessario alcun aggiustamento della dose; Grado 3 (da> 3 a ≤10 x ULN): utilizzare il preparato se la concentrazione di bilirubina diminuisce al grado ≤ 1 (da> ULN a 1,5 x ULN), quindi ridurre la dose (vedere lo schema di riduzione della dose); Grado 4 (> 10 x ULN): interrompere il trattamento. Regole di modifica della dose per trombocitopenia: grado 3 (concentrazione piastrinica da 25.000 a 3) - utilizzare il preparato quando la concentrazione piastrinica è ≤1. (ad esempio, ≥75.000 / mm3); nessun aggiustamento della dose necessario; Grado 4 (concentrazione piastrinica 3): utilizzare il preparato quando la concentrazione piastrinica raggiunge ≤1. (ad es. ≥75.000 / mm3) quindi ridurre la dose (vedere lo schema di riduzione della dose). Principi di modifica della dose in pazienti con disfunzione ventricolare: LVEF 45% - continuare la terapia; LVEF dal 40% al ≤45% con contemporanea riduzione della frazione di eiezione di - non utilizzare il preparato; Rivalutare la LVEF entro 3 settimane, se la LVEF è ancora ≥10 punti percentuali rispetto al basale, interrompere il trattamento; CHF sintomatico - interrompere il trattamento. Bambini e adolescenti: la sicurezza e l'efficacia nei bambini e negli adolescenti di età inferiore a 18 anni non sono state stabilite poiché il carcinoma mammario disseminato (MBC) non è noto in questa popolazione. Gruppi speciali di pazienti. La terapia deve essere temporaneamente interrotta nei pazienti che sviluppano neuropatia periferica di Grado 3 o 4 fino a raggiungere il Grado ≤2; quando si riprende il trattamento, si può prendere in considerazione la riduzione della dose come delineato nello schema di riduzione della dose. Non è richiesto alcun aggiustamento della dose per i pazienti di età ≥ 65 anni; non ci sono dati sufficienti per determinare la sicurezza e l'efficacia della terapia in pazienti di età ≥75 anni. Non è richiesto alcun aggiustamento della dose iniziale nei pazienti con insufficienza renale lieve o moderata; La potenziale necessità di aggiustamento della dose nei pazienti con grave insufficienza renale non può essere determinata e pertanto questi pazienti devono essere attentamente monitorati. Non è richiesto alcun aggiustamento della dose iniziale nei pazienti con insufficienza epatica lieve o moderata. Trastuzumab emtansine non è stato studiato in pazienti con grave insufficienza epatica; si deve usare cautela nel trattamento di pazienti con compromissione epatica a causa della nota epatotossicità osservata durante il trattamento. Modo di dare. Il preparato deve essere sciolto e diluito da personale medico e somministrato come infusione endovenosa. Non deve essere somministrato come bolo o iniezione rapida.
Indicazioni
Monoterapia di pazienti adulti con carcinoma mammario localmente avanzato o metastatico HER2 positivo, inoperabile, precedentemente trattati con trastuzumab e un taxano, in associazione o separatamente. Pazienti che sono stati precedentemente sottoposti a trattamento per malattia localmente avanzata o generalizzata o che hanno avuto una recidiva durante o entro 6 mesi dal completamento della terapia adiuvante.
Controindicazioni
Ipersensibilità al principio attivo o ad altri ingredienti del preparato.
Precauzioni
Negli studi clinici con trastuzumab emtansine sono stati segnalati casi di malattia polmonare interstiziale (ILD), inclusa la polmonite; alcuni sono stati associati a insufficienza respiratoria acuta o sono stati fatali. Si raccomanda l'interruzione del trattamento con il preparato nei pazienti con diagnosi di ILD o polmonite. I pazienti con dispnea a riposo, associata a complicanze del cancro avanzato e malattie associate possono essere maggiormente a rischio di sviluppare complicanze polmonari. Durante il trattamento con il preparato, sono stati osservati epatotossicità e gravi disturbi del fegato e delle vie biliari, inclusa l'ipertrofia rigenerativa nodulare (NRH) del fegato e decessi dovuti a danno epatico indotto da farmaci; possono essere stati influenzati anche comorbidità e / o farmaci concomitanti noti per avere un potenziale epatotossico. La funzionalità epatica deve essere monitorata prima dell'inizio del trattamento e prima del dosaggio successivo. I pazienti con un aumento delle ALT al basale (ad es. Associato a metastasi epatiche) sono predisposti allo sviluppo di insufficienza epatica e sono a più alto rischio di tossicità epatica di Grado 3-5 o livelli di laboratorio epatici elevati. I casi di NRH sono stati diagnosticati mediante biopsie epatiche. La presenza di NRH deve essere considerata in tutti i pazienti con segni clinici di ipertensione portale e / o immagine simile alla cirrosi osservata alla tomografia computerizzata epatica ma con livelli sierici normali di transaminasi e nessun'altra evidenza di cirrosi. Se viene diagnosticata NRH, il trattamento con il preparato deve essere interrotto. Non è stato studiato in pazienti con transaminasi sieriche> 2,5 x ULN (limite superiore della norma) o con bilirubina totale> 1,5 x ULN prima dell'inizio del trattamento. In caso di attività delle transaminasi sieriche> 3 x ULN e contemporaneamente concentrazione di bilirubina totale> 2 x ULN, il trattamento deve essere interrotto. Si deve usare cautela nel trattamento di pazienti con compromissione epatica. Vi è un aumento del rischio di disfunzione ventricolare sinistra durante il trattamento. Nei pazienti trattati con trastuzumab emtansine, è stata osservata una riduzione della frazione di eiezione ventricolare sinistra (LVEF) a 50 anni), con un valore iniziale di LVEF basso (25 kg / m2). I test di funzionalità cardiaca standard (ecocardiogramma o angiografia con radionuclidi multi-gate) devono essere eseguiti prima dell'inizio del trattamento ea intervalli regolari (ad esempio ogni 3 mesi). La LVEF dei pazienti al basale era ≥50% nella maggior parte degli studi clinici. Sono stati esclusi dallo studio i pazienti con insufficienza cardiaca congestizia, aritmie gravi che richiedono un trattamento, anamnesi di infarto miocardico o angina instabile nei 6 mesi precedenti la randomizzazione o con dispnea a riposo dovuta a cancro avanzato. In caso di disturbi del ventricolo sinistro, la somministrazione della dose successiva deve essere posticipata o la terapia deve essere interrotta. L'effetto del trattamento con trastuzumab emtansine nei pazienti che interrompono il trattamento con trastuzumab a causa di una reazione correlata all'infusione non è stato studiato; la terapia con questo farmaco non è raccomandata in questo gruppo di pazienti. La terapia deve essere interrotta nei pazienti che sviluppano una grave reazione correlata all'infusione fino alla risoluzione dei sintomi. La ripresa della terapia deve essere presa in considerazione sulla base del giudizio clinico sulla gravità della reazione. Il trattamento deve essere interrotto in caso di reazione all'infusione pericolosa per la vita. L'effetto del trattamento con trastuzumab emtansine nei pazienti che hanno interrotto il trattamento con trastuzumab a causa dell'ipersensibilità non è stato studiato; L'uso di trastuzumab emtansine in questi pazienti non è raccomandato. I pazienti devono essere monitorati per ipersensibilità / reazioni allergiche, i sintomi possono essere gli stessi delle reazioni correlate all'infusione; sono state osservate gravi reazioni anafilattiche. Se si sviluppa ipersensibilità (con aumento delle reazioni durante le infusioni successive), il trattamento con trastuzumab emtansine deve essere interrotto.A causa del rischio di eventi emorragici (inclusa emorragia del SNC, respiratoria e gastrointestinale), è necessario prestare attenzione e prendere in considerazione un monitoraggio aggiuntivo in caso di somministrazione concomitante con anticoagulanti o farmaci antipiastrinici. Si raccomanda di monitorare la conta piastrinica prima della somministrazione di ciascuna dose di trastuzumab emtansine. I pazienti con trombocitopenia (≤100.000 / mm3) e i pazienti in terapia anticoagulante (ad es. Warfarin, eparina, eparine a basso peso molecolare) devono essere attentamente monitorati durante il trattamento con trastuzumab emtansine. Trastuzumab emtansine non è stato studiato in pazienti con conta piastrinica ≤100.000 / mm3 prima dell'inizio della terapia. In caso di peggioramento della trombocitopenia al Grado 3 o superiore (3), il preparato non deve essere utilizzato fino a quando la tossicità non scende al Grado 1 (≥75.000 / mm3). Negli studi clinici con trastuzumab emtansine si è verificata una neuropatia periferica di grado 1, principalmente sensoriale. I pazienti con neuropatia periferica di Grado ≥3 sono stati esclusi dalla partecipazione a studi clinici. Il trattamento dei pazienti con neuropatia periferica di grado 3 o 4 deve essere periodicamente interrotto fino a quando la complicanza non si riduce a Grado ≤2. I pazienti devono essere monitorati regolarmente per segni di neurotossicità. Al fine di migliorare la tracciabilità dei preparati biologici, il nome commerciale del farmaco somministrato deve essere chiaramente scritto (o dichiarato) nella cartella del paziente. La sicurezza e l'efficacia del farmaco nei bambini e negli adolescenti di età inferiore ai 18 anni non sono state stabilite poiché il cancro al seno disseminato non è stato riscontrato in questa popolazione. Non deve essere somministrato come bolo o iniezione rapida.
Attività indesiderabile
Molto comune: infezione del tratto urinario, trombocitopenia, anemia, ipopotassiemia, insonnia, neuropatia periferica, mal di testa, sanguinamento, epistassi, tosse, dispnea, stomatite, diarrea, vomito, nausea, costipazione, bocca secca, dolore addominale , eruzione cutanea, dolore muscoloscheletrico, artralgia, mialgia, affaticamento, febbre, astenia, brividi, aumento delle transaminasi. Comune: neutropenia, leucopenia, ipersensibilità, capogiri, disgeusia, disturbi della memoria, sindrome dell'occhio secco, congiuntivite, disturbi visivi, aumento della lacrimazione, disfunzione ventricolare sinistra, ipertensione, dispepsia, sanguinamento gengivale, prurito, alopecia, malattia delle unghie , eritrodisestesia palmo-plantare, orticaria, edema periferico, aumento della fosfatasi alcalina, reazioni all'infusione (arrossamento della pelle, brividi, febbre, dispnea, pressione sanguigna bassa, respiro sibilante, broncospasmo, tachicardia). Non comune: polmonite (ILD), epatotossicità, insufficienza epatica, ipertrofia rigenerativa nodulare, ipertensione portale, stravaso nel sito di iniezione (eritema, dolorabilità, irritazione cutanea, dolore o gonfiore). Inoltre, è stata osservata iperbilirubinemia. Negli studi clinici, aumenti delle transaminasi sieriche (Grado 1-4) si sono verificati durante il trattamento con trastuzumab emtansine ed erano generalmente transitori. L'aumento delle transaminasi è stato più spesso transitorio, con un picco all'ottavo giorno dopo la somministrazione. Successivamente, la tossicità è diminuita al Grado 1 o si è risolta prima del ciclo successivo. Si è verificato anche un effetto cumulativo (la percentuale di pazienti con innalzamento delle ALT / AST di Grado 1-2 è aumentata con i cicli successivi). Nei pazienti con livelli elevati di transaminasi, la maggior parte dei pazienti ha manifestato una riduzione o un recupero della tossicità di Grado 1 entro 30 giorni dall'ultima dose di trastuzumab emtansine. La disfunzione ventricolare sinistra è stata riscontrata nel 2,2% dei pazienti che hanno partecipato a studi clinici. Nella maggior parte dei casi, le diminuzioni di LVEF di grado 1 o 2 erano asintomatiche e la tossicità di grado 3 o 4 è stata osservata nello 0,4% dei pazienti, di solito durante i cicli iniziali di trattamento (1-2). Eventi emorragici gravi (Grado ≥3) sono stati riportati nel 2,2% di tutti i pazienti. La trombocitopenia si è verificata nel 24,9% dei pazienti negli studi clinici ed è stata la reazione avversa più comune che ha portato all'interruzione della terapia. Negli studi clinici, l'incidenza e la gravità della trombocitopenia erano maggiori nei pazienti asiatici. Alcuni pazienti che hanno sviluppato queste complicanze sono stati trattati anche con anticoagulanti. Si sono verificati episodi emorragici fatali e gravi complicanze emorragiche, incluso sanguinamento del SNC. Il 5,3% dei pazienti è risultato positivo agli anticorpi anti-trastuzumab emtansine.
Gravidanza e allattamento
L'uso di trastuzumab emtansine nelle donne in gravidanza non è raccomandato. Prima di iniziare una gravidanza, le donne devono essere informate della possibilità di danni al feto. Tuttavia, le pazienti che rimangono incinte devono contattare immediatamente un medico. Se una donna incinta è trattata con trastuzumab emtansine, si raccomanda uno stretto monitoraggio da parte di un team multidisciplinare. Trastuzumab può causare danni al feto o morte se somministrato a una donna incinta. Nei bambini di donne in gravidanza trattate con il preparato, sono stati riportati casi di oligoidramnios, alcuni dei quali hanno portato a ipoplasia polmonare fatale. DM1, il componente citotossico di trastuzumab emtansine, può essere teratogeno e potenzialmente embriotossico. Le donne devono interrompere l'allattamento al seno prima di iniziare la terapia con trastuzumab emtansine. I pazienti possono iniziare l'allattamento al seno 7 mesi dopo l'interruzione del trattamento. Le donne in età fertile devono usare misure contraccettive efficaci durante il trattamento con il farmaco e per 7 mesi dopo l'ultima dose di trastuzumab emtansine. Anche gli uomini o le loro partner dovrebbero utilizzare metodi contraccettivi efficaci.
Commenti
I pazienti che manifestano reazioni correlate all'infusione non devono guidare o usare macchinari fino alla risoluzione di questi sintomi. Informazioni redatte sulla base di SPC di 13 luglio 2017 L'attuale RCP è disponibile su www.roche.pl.
Interazioni
I risultati in vitro degli studi sul metabolismo nei microsomi epatici umani indicano che DM1 è metabolizzato principalmente dall'enzima CYP3A4 e in misura minore dal CYP3A5. L'uso concomitante di potenti inibitori del CYP3A4 (ad es. Ketoconazolo, itraconazolo, claritromicina, atazanavir, indinavir, nefazodone, nelfinavir, ritonavir, saquinavir, telitromicina e voriconazolo) deve essere evitato poiché ciò può aumentare i livelli di DM1 e la tossicità. Si dovrebbe prendere in considerazione una formulazione alternativa che non inibisca o inibisca solo leggermente il CYP3A4. Se l'uso concomitante di potenti inibitori del CYP3A4 non può essere evitato e, ove possibile, considerare di ritardare la somministrazione di trastuzumab emtansine fino a quando gli inibitori del CYP3A4 non vengono eliminati dalla circolazione (circa 3 emivite degli inibitori). Tuttavia, se viene usato contemporaneamente un potente inibitore del CYP3A4 e il trattamento con trastuzumab emtansine non può essere ritardato, il paziente deve essere attentamente monitorato per gli effetti avversi in questi casi.
Il preparato contiene la sostanza: Trastuzumab emtansine
Droga rimborsata: NO
















-budowa-i-funkcje.jpg)



